
Overview
Autoimmune encephalitis (AE) is inflammation of the brain caused by immune system dysfunction. It is an umbrella term for a heterogenous group of inflammatory brain disorders that are non-infectious and immune-mediated. Onset can be acute or subacute and commonly manifests as a combination of:
- psychiatric symptoms e.g. hallucinations, anxiety, mania, depression, agitation, personality changes;
- cognitive disturbances e.g. memory loss, confusion, impaired communication; and/or
- seizures.
CAUSEs
In broad terms, autoimmune encephalitis (AE) occurs when a person's own immune system mounts a defense against parts of their own brain. The trigger for this autoimmune cascade could be:
As for the resulting inflammation, this can be attributed to a pathogenic autoantibody in ~50% of cases. It was only in 2007 that Dr. Josep Dalmau and colleagues at the University of Pennsylvania identified the first autoantibody: the NMDA receptor autoantibody, which causes the most common subtype of AE. Since then, dozens have been described and the list of AE-associated autoantibodies grows by the year. But many more remain to be elucidated. Patients without an identifiable autoantibody often have a delayed diagnosis, suboptimal treatment, and worse outcomes. That's part of why supporting ongoing AE research remains so important.
- a tumor e.g. a teratoma
- exposure to a pathogen e.g. herpes simplex virus
- specialised immune-modulating medications e.g. immune check point inhibitors
As for the resulting inflammation, this can be attributed to a pathogenic autoantibody in ~50% of cases. It was only in 2007 that Dr. Josep Dalmau and colleagues at the University of Pennsylvania identified the first autoantibody: the NMDA receptor autoantibody, which causes the most common subtype of AE. Since then, dozens have been described and the list of AE-associated autoantibodies grows by the year. But many more remain to be elucidated. Patients without an identifiable autoantibody often have a delayed diagnosis, suboptimal treatment, and worse outcomes. That's part of why supporting ongoing AE research remains so important.
classification schemes
There are a few different classification schemes used to subdivide the autoimmune encephalitides. They can be categorized according to their:
- anatomical classification i.e. location of inflammation within the nervous system
- serological classification i.e. antibody identified in serum and/or cerebrospinal fluid
- etiological classification i.e. cause
- degree of diagnostic certainty i.e. possible, probable, or definite
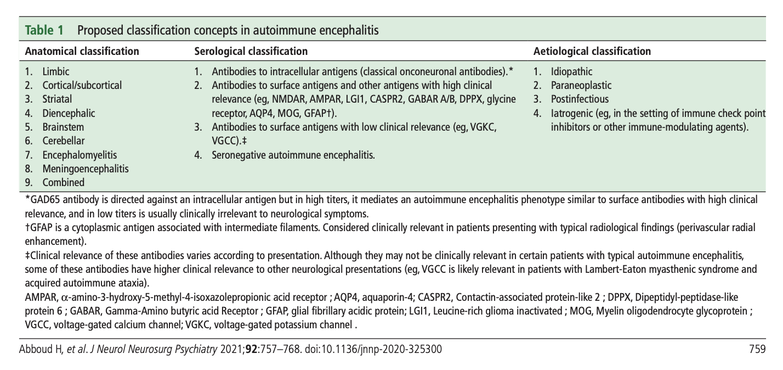
In practice, doctors primarily use the serological classification scheme and refer to a case according to the antibody identified. In cases where no antibody is identified, patients are referred to as having seronegative AE.
If a patient with seronegative AE has evidence of limbic system inflammation they are referred to as having limbic encephalitis, as this is better defined than seronegative AE (which encompasses a wider spectrum of autoimmune encephalitides). But sometimes doctors use the term limbic encephalitis loosely, as it was the diagnostic label used historically for decades before the advent of autoantibody discovery.
If a patient with seronegative AE has evidence of limbic system inflammation they are referred to as having limbic encephalitis, as this is better defined than seronegative AE (which encompasses a wider spectrum of autoimmune encephalitides). But sometimes doctors use the term limbic encephalitis loosely, as it was the diagnostic label used historically for decades before the advent of autoantibody discovery.
select autoimmune encephalitis subtypes
LIMBIC ENCEPHALITIS
This group of encephalitides is characterized by inflammation of the limbic system. The most telling sign of limbic encephalitis is severe impairment of short-term memory and seizures. Other symptoms may include confusion, psychiatric symptoms, and seizures. The symptoms typically develop over a few weeks or months, but in some cases they may evolve over just a few days. Limbic encephalitis is often associated with an underlying neoplasm (paraneoplastic limbic encephalitis); however some cases never have a neoplasm identified (non-paraneoplastic limbic encephalitis).
This group of encephalitides is characterized by inflammation of the limbic system. The most telling sign of limbic encephalitis is severe impairment of short-term memory and seizures. Other symptoms may include confusion, psychiatric symptoms, and seizures. The symptoms typically develop over a few weeks or months, but in some cases they may evolve over just a few days. Limbic encephalitis is often associated with an underlying neoplasm (paraneoplastic limbic encephalitis); however some cases never have a neoplasm identified (non-paraneoplastic limbic encephalitis).
|
ANTI-N-METHYL-D-ASPARTATE RECEPTOR (NMDAR) ENCEPHALITIS
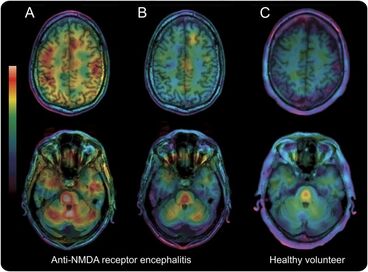
Anti-NMDAR encephalitis was the first antibody-mediated encephalitis discovered and is also the most common. It occurs when antibodies produced by the body’s own immune system attack NMDA receptors in the brain. Anti-NMDA patients typically have a dramatic presentation because the NMDA receptors are vital for functions such as memory, behavior, and perception of reality. Before they receive the diagnosis, many patients are thought to be drunk, on drugs, or psychotic due to the way they interact with others, the paranoia they feel, and the hallucinations they experience. Anti-NMDA receptor encephalitis is most common in young women and is often associated with an ovarian teratoma. Less commonly it can occur in males in association with a testicular teratoma. One pathognomic sign is the appearance of a delta brush on EEG studies. |
|
LEUCINE-RICH GLIOMA INACTIVATED PROTEIN (LGI1)-ANTIBODY ENCEPHALITIS
LGI1-antibody encephalitis is the second most common form of AE and typically manifests as subacute memory impairment, seizures, and mental and behaviour abnormalities. It is most often found in elderly males. One pathognomic sign is a distinct form of seizure known as faciobrachial dystonic seizures.
LGI1-antibody encephalitis is the second most common form of AE and typically manifests as subacute memory impairment, seizures, and mental and behaviour abnormalities. It is most often found in elderly males. One pathognomic sign is a distinct form of seizure known as faciobrachial dystonic seizures.
ANTI-GABA AMINOBUTYRIC ACID B RECEPTOR (GABABR) ENCEPHALITIS
Anti-GABABR encephalitis typically manifests as acute memory impairment and seizures and is often found in association with lung cancer. On average it has a worse prognosis than other AE subtypes, with higher mortality rates. It typically impacts elderly patient populations.
Anti-GABABR encephalitis typically manifests as acute memory impairment and seizures and is often found in association with lung cancer. On average it has a worse prognosis than other AE subtypes, with higher mortality rates. It typically impacts elderly patient populations.
CONTACTIN-ASSOCIATED PROTEIN 2 (CASPR2)-ANTIBODY ENCEPHALITIS
CASPR2-antibody encephalitis can have a more chronic evolution compared with other AE subtypes. It typically impacts elderly males.
CASPR2-antibody encephalitis can have a more chronic evolution compared with other AE subtypes. It typically impacts elderly males.
MYELIN-OLIGODENDROCYTE GLYCOPROTEIN (MOG)-ANTIBODY ENCEPHALOMYELITIS
MOG-antibody encephalomyelitis can present as acute disseminated encephalomyelitis (ADEM) with optic neuritis and transverse myelitis. It typically impacts younger patient populations.
MOG-antibody encephalomyelitis can present as acute disseminated encephalomyelitis (ADEM) with optic neuritis and transverse myelitis. It typically impacts younger patient populations.
ANTI-GLUTAMIC ACID DECARBOXYLASE (GAD65) ENCEPHALITIS
Anti-GAD65 antibodies are classically associated with three syndromes: stiff person syndrome, cerebellar ataxia, and limbic encephalitis. It can also occur in patients with type 1 diabetes mellitus. Anti-GAD65 encephalitis is associated with high serum titres of anti-GAD65 antibodies, as low serum titres can be found in the general population and may be considered non-specific.
Unlike most other identified nonparaneoplastic autoantibodies which target the neuronal surface, anti-GAD65 antibodies target intracellular antigens. Whether or not it is directly responsible for the pathogenesis of encephalitis remains uncertain. The average patient tends not to be as responsive to immunotherapies as those with other types of AE.
Anti-GAD65 antibodies are classically associated with three syndromes: stiff person syndrome, cerebellar ataxia, and limbic encephalitis. It can also occur in patients with type 1 diabetes mellitus. Anti-GAD65 encephalitis is associated with high serum titres of anti-GAD65 antibodies, as low serum titres can be found in the general population and may be considered non-specific.
Unlike most other identified nonparaneoplastic autoantibodies which target the neuronal surface, anti-GAD65 antibodies target intracellular antigens. Whether or not it is directly responsible for the pathogenesis of encephalitis remains uncertain. The average patient tends not to be as responsive to immunotherapies as those with other types of AE.
SERONEGATIVE AUTOIMMUNE ENCEPHALITIS
Approximately 50% of AE cases are seronegative, meaning that an autoantibody has not been identified in the patient's serum or CSF. This could be for a number of reasons. Perhaps scientists have not yet discovered the antibody responsible for causing the encephalitis, the titre of antibodies is below the test's threshold of sensitivity, or the encephalitis is not antibody-mediated but rather T-cell mediated.
Without an identifiable autoantibody, patients with seronegative AE endure particular hardship in order to receive a diagnosis. They are one of the most underserved AE populations, with high rates of misdiagnosis, excessive treatment delays, and insurance barriers. The result is often worse recovery outcomes. However, studies have shown that the average seronegative AE patient has a high potential for recovery if diagnosed and treated in a timely manner.
Approximately 50% of AE cases are seronegative, meaning that an autoantibody has not been identified in the patient's serum or CSF. This could be for a number of reasons. Perhaps scientists have not yet discovered the antibody responsible for causing the encephalitis, the titre of antibodies is below the test's threshold of sensitivity, or the encephalitis is not antibody-mediated but rather T-cell mediated.
Without an identifiable autoantibody, patients with seronegative AE endure particular hardship in order to receive a diagnosis. They are one of the most underserved AE populations, with high rates of misdiagnosis, excessive treatment delays, and insurance barriers. The result is often worse recovery outcomes. However, studies have shown that the average seronegative AE patient has a high potential for recovery if diagnosed and treated in a timely manner.

other forms of encephalitis with suspected autoimmune involvement
HASHIMOTO'S ENCEPHALOPATHY (HE)/STEROID RESPONSIVE ENCEPHALOPATHY ASSOCIATED WITH THYROIDITIS (SREAT)
Hashimoto's encephalopathy is a condition characterized by onset of confusion with altered level of consciousness, seizures, jerking of muscles (myoclonus), and sometimes psychosis including visual hallucinations and paranoid delusions. It is often found in association with elevated thyroid antibodies. Most HE patients respond well to steroids, hence the alternate name of steroid responsive encephalopathy associated with thyroiditis (SREAT).
Hashimoto's encephalopathy is a condition characterized by onset of confusion with altered level of consciousness, seizures, jerking of muscles (myoclonus), and sometimes psychosis including visual hallucinations and paranoid delusions. It is often found in association with elevated thyroid antibodies. Most HE patients respond well to steroids, hence the alternate name of steroid responsive encephalopathy associated with thyroiditis (SREAT).
RASMUSSEN'S ENCEPHALITIS
Rasmussen's encephalitis is a progressive and chronic form of brain inflammation that typically affects only one hemisphere of the brain. It most often occurs in children and causes severe epileptic activity. The result is permanent brain damage, loss of motor skills and speech, paralysis on one side of the body (hemiparesis), and mental deterioration. An estimated 200-500 cases exist worldwide, making it extremely rare.
Rasmussen's encephalitis is a progressive and chronic form of brain inflammation that typically affects only one hemisphere of the brain. It most often occurs in children and causes severe epileptic activity. The result is permanent brain damage, loss of motor skills and speech, paralysis on one side of the body (hemiparesis), and mental deterioration. An estimated 200-500 cases exist worldwide, making it extremely rare.
Other RElated neurological illnesses
ACUTE DISSEMINATED ENCEPHALOMYELITIS (ADEM)
Acute disseminated encephalomyelitis (ADEM) is a rare neurological disorder that causes inflammation of the brain and spinal cord. It is thought to occur after a trigger (usually a viral or bacterial illness; sometimes a vaccination) causes an abnormal immune system response. The result is demyelination (damage to the myelin sheath) and neurological symptoms that presents within days to weeks. In some cases it is mild and self-limited; in other cases it causes life-threatening complications.
Acute disseminated encephalomyelitis (ADEM) is a rare neurological disorder that causes inflammation of the brain and spinal cord. It is thought to occur after a trigger (usually a viral or bacterial illness; sometimes a vaccination) causes an abnormal immune system response. The result is demyelination (damage to the myelin sheath) and neurological symptoms that presents within days to weeks. In some cases it is mild and self-limited; in other cases it causes life-threatening complications.